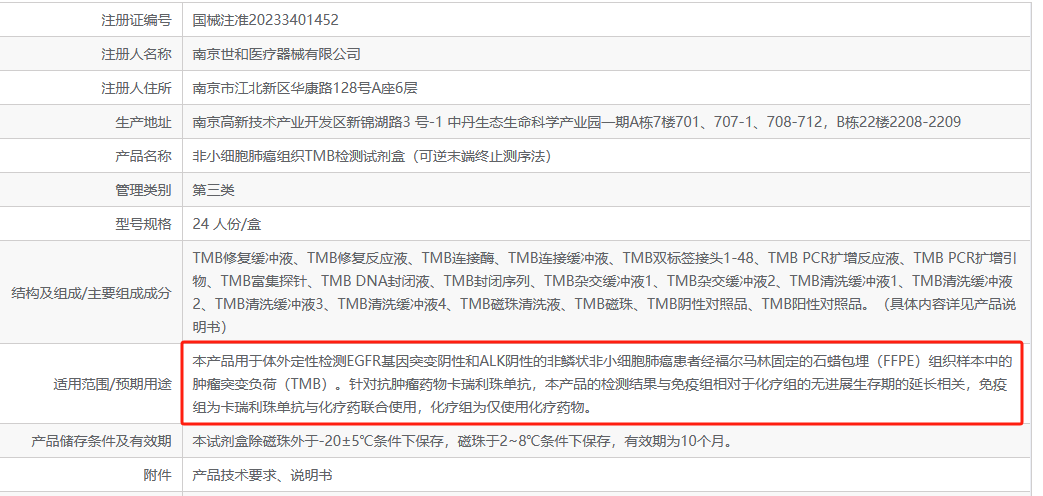
谋士以身做饵,诱天下人入局——前言10月12日,世和的TMB试剂盒获批毫无疑问是肿瘤NGS圈今年最炸裂的新闻之一。随之而来的,是铺天盖地的“质疑” ...
|
谋士以身做饵,诱天下人入局
10月12日,世和的TMB 试剂盒获批毫无疑问是肿瘤 NGS 圈今年最炸裂的新闻之一。 随之而来的,是铺天盖地的“质疑”,因为这个证挑战了大家“长期默认”的大panel注册路径。
可以复制吗?如何复制? 注册审评报告无疑成了所有人期待的那个“答案”。 不过,似乎这个审评报告并不会那么快到来——从历史数据看,审评报告平均的上线时间是产品获批后的 40 天——也有极个别创新医疗器械一直没能等到自己的审评报告。 上一篇文章写完,其实我脑海里一直回响的是两位大佬的话: “就是注册角度来看,还是那个观点哈,只要有坚定意志推的产品,一定可以拿个证的。终究可以拿个证的。不管是多么不明朗的监管。” “如果你不挑三拣四,那我就给你指一条路,你走得通就给你,走不通别怪我了。至于这条路,和初衷是如何天差地别,和研究趋势有多么不同,已经不重要了” 在审评报告出来之前,谨以我浅薄的认知,尝试推测大panel的注册路径,去回答以下四个问题,也算是上一篇《肿瘤 NGS大panel 第一证:勇气、疑问和影响》在注册路径方向的延展:
PS:这篇所有引用材料均来自公开信息,注册实在是一门太深的学问,如有理解不对的地方欢迎留言指教、指正。 01 “大Panel第一证”可以是什么 这个问题换个问法是:TMB的临床意义可以是什么? 临床意义本身一定是一个检测的必备要素,就像 Z 老师说的那句话: “医疗器械产品,是有定义的,你检测的准,只是个产品,不是医疗器械” 关于临床意义可以结合两个参考材料来看,一份来自CDE
其中将生物标志物分成了六大类: 诊断性、预后性、预测性、药效学、安全性、检测性 其中TMB 只可能是两类,分别定义如下
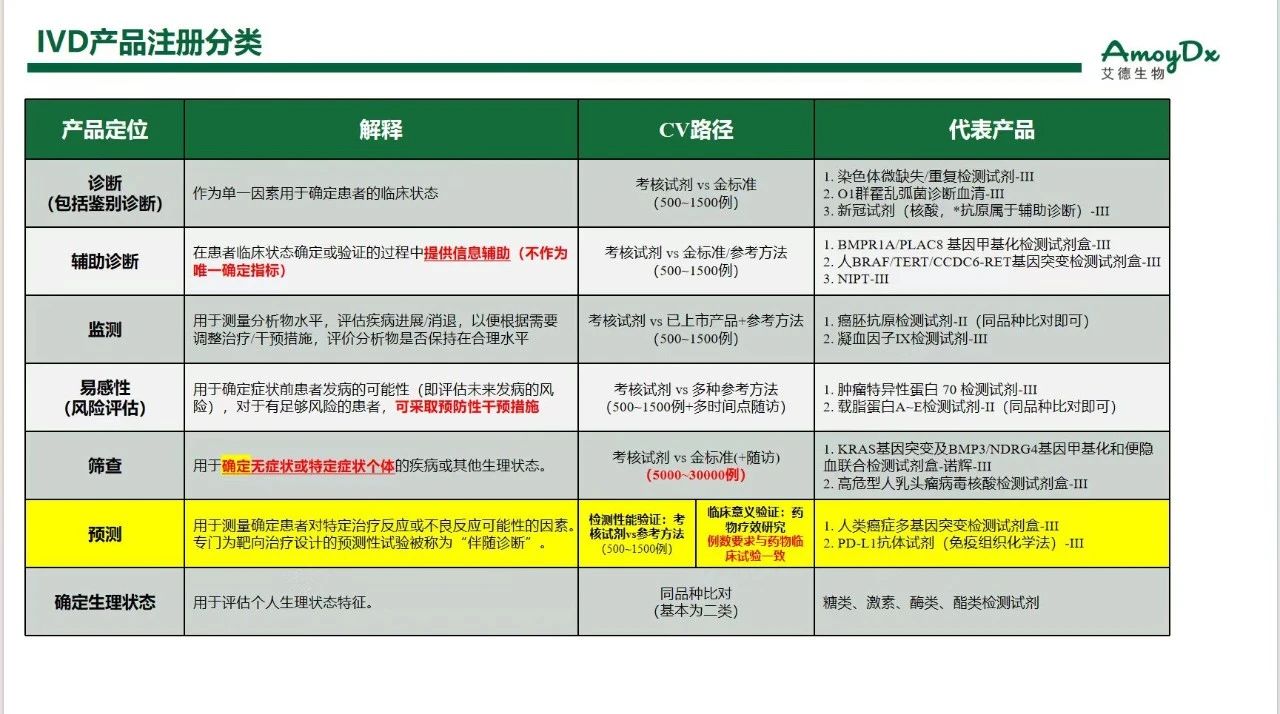
另一份参考材料是CMDE的培训,关于 IVD 产品注册分类(自己理解整理的,不保证理解完全正确)
考虑到CMDE和CDE毕竟是平行部门而非从属关系,二者之间的对应关系并没有官方说法,但从常规理解,我们可以这么对应:
那么大 Panel 第一证更可能是哪个呢? 02 为什么大概率不是伴随诊断 顺带也回答下为什么大概率不是补充诊断? 还是从CDE和CMDE两个视角来分析这个问题。大Panel第一证此次是有做药效研究的,这个研究是卡瑞利珠单的注册临床试验CameL。 在 CMDE 关于IVD产品注册分类的培训材料中,有一条间接否定了其作为伴随诊断:
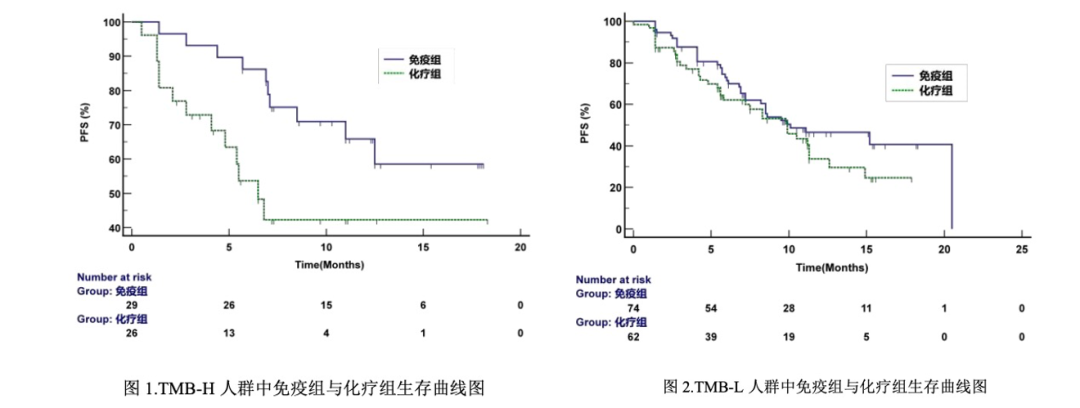
而CameL入组了病例 412 例,但能够被用于该试剂盒药效研究的只剩下 218 例(有效 TMB 结果 192 例)。
而根据CDE的《生物标志物在抗肿瘤药物临床研发中应用的技术指导原则》亦有两条可以直接否定其作为伴随诊断:
退一步说,至少 TMB 肯定不能被归类为一级位点(CMDE 9月份的公众号小作文,《基于高通量测序技术(NGS)的肿瘤基因变异检测伴随诊断试剂的检测范围可以包括哪些基因及位点?》)
而一个伴随诊断试剂盒是不应该只有二级位点而没一级位点的。 这里遗留了一个问题,那就是“补充诊断”,虽然官方一直没有明确的定义,但从既往获批的补充诊断试剂盒(Dako 的 PD-L1) 来看,是可以“不反映在药物说明书中”+“整体人群获益而非只有 M+人群获益”的。 而且这个 PD-L1 的预期用途描述实在和大 Panel 第一证太像了,很难不让人产生联想:
那么,大Panel 第一证可以是补充诊断吗? 先看下PD-L1 的补充诊断是如何获批的,因从其注册审评报告中有其参与纳武单抗中国注册临床 Checkmate078 的完整数据,可以大胆推测一下“补充诊断”的一个必备要素:
而在CameL 研究中,并不存在这样一个亚组,因此大 panel 第一证也不太可能是补充诊断。 03 可能是“辅助诊断”吗 “没有说必须是伴随诊断,同样是计算得到的指标MSI,国内批的也不是伴随诊断,是辅助诊断” 这句话来自K老师,用于回答我关于“TMB 天然必须得是伴随诊断吗?”的疑问。国内目前获批的三个MSI试剂盒在预期用途里都有这么一句话:
那么大panel 第一证的临床意义可以仅仅是辅助诊断TMB-H的非鳞状非小细胞肺癌吗? 关于 MSI我们可以参考的最权威资料当然是这个:
其中明确提到 MSI 的临床意义,可以总结如下:
因此只要 MSI DNA 检测能够证实自己和金标准的一致性“与MMR蛋白IHC检测的阳性符合率、阴性符合率95%置信区间下限一般分别不低于90%和95%”,就可以为dMMR 这种临床状态确定提供信息辅助。 这是MSI 试剂盒能够以“辅助诊断”的产品定位获批的前提条件。 如果按照这个逻辑推断,只要能够满足:
那么 TMB 试剂盒就也可以以“辅助诊断”的产品定位获批。从目前的公开信息看,这两个问题有一半是“已知”的:
剩下的另一半是,TMB-H的金标准是什么? 在《肿瘤组织基因突变检测试剂盒(高通量测序法)》的国标征求意见稿中,明确提到需要依据“药效数据”来区分 TMB-H/L 。
从这个角度来看,这个试剂盒参与CameL 后筛选出的192 例TMB 检测人群可以看做是一个“独立”的临床试验——一个不是出于伴随诊断目的,而仅仅出于验证TMB 阳性判断值的临床试验。 这倒也侧面 echo 了一位朋友在某次CIMDR上咨询监管机构人士得到的信息: “TMB不能只做准确性临床来申报,要和药物一起做临床试验……也没说必须是伴随诊断的方式……国内企业要和药物一起做临床,怎么做自己想办法” 从目前能获得的信息看,这个验证从结果中看倒是没什么问题,验证了其“TMB 状态的判定标准:当 TMB 值≥10 个/Mb 时,TMB 状态为 高 T M B ( T M B - H ); 当 T M B 值 < 1 0 个 / M b 时 , T M B 状 态 为 低 T M B (TMB-L)”的阳性判断值是成立的。
综合来看,“辅助诊断”应该是这张大panel 第一证最有可能的产品定位。 当然,这里还有个问题无法推测,需要等待审评报告: 在和药物一起做临床的时候,如果可以从中筛选患者形成一个“独立”的临床试验,这个筛选是否有明确的规则要求或者限制?比如这次直接筛选到只剩药物临床试验的不到 50%似乎也有点夸张了。 04 未来会怎么样 如果大 panel 第一证是“辅助诊断”的推断是正确的,可以解答一个疑惑,也会对行业的未来产生巨大的影响。
因为这样就跨注册单元了,这在当前的 IVD 产品注册实践中是个尚未被突破的事情。“辅助诊断”和“伴随诊断”这两种截然不同的“临床预期用途”应该无法被归类为同一注册单元。(A 司那个产品大概是个“例外/意外”,后无来者是有原因的)。 在《医疗器械注册单元划分指导原则》中有如下描述
准确的说,如果只是为了拿证,将变得“简单”。 这里可以参考下 《微卫星不稳定性(MSI)检测试剂临床评价注册审查指导原则》,在已经有同类产品获批的情况下,虽然各产品在 TMB位点选择、算法上都不同,但应该都可以类比 MSI 中的“如果申报产品采用全新的微卫星位点组合(包括全新的MSI-H判定标准),该组合在以往国内、外权威指南、规范等文件中均未有推荐,且尚未受到行业广泛认可”通过如下三步完成临床性能评价
1. 与 WES TMB 阳性/阴性符合率的一致性比较研究。 2. 与已上市同类产品进行对比试验,评价两种试剂判定 TMB-H和TMB-L的一致性。 3. 与 WES TMB针对特定基因的突变位点进行一致性比较研究。 省掉了最麻烦的药效临床试验,相比之前不是容易了一星半点。 不过 TMB 的注册审查指导原则应该不会这么快出来,因为似乎 CMDE 有一个标准:必须有5个以上的产品有案例(不一定是获批,主要是能够形成一个标准流程进行审查了,可以是某个产品没达到这个标准要求因此无法获批),没有5个案例是不允许起草/发布注册指导原则的。
2018年燃石的NGS小panel 第一证迈出了了肿瘤 NGS 合规化的第一步,在这之后大家依然做了大量的探索,包括扩展癌种、扩展突变类型、桥接伴随诊断等等。 如果这一次 世和的大panel 第一证是“辅助诊断”的话,留给大家去探索的空间还有无限大,比如伴随诊断、突破单癌种等等。 这,是一个起点,而非终点。 05 结尾 落笔最后一个字的时候,黎明将至,东方既白。 就像一片光明的未来,向着肿瘤 NGS 行业张开怀抱。 |


微信公众号

微博账号

商务合作