今年下半年,业内关于“规范基因检测外送”、“医疗反腐”的消息不绝于耳,整个基因检测行业式微的讨论甚嚣尘上。10月以来,世和TMB、艾德MSI获批的消息先后刷屏,给行业注入了两剂“强心针”,一扫眼下低迷之势。前 ...
|
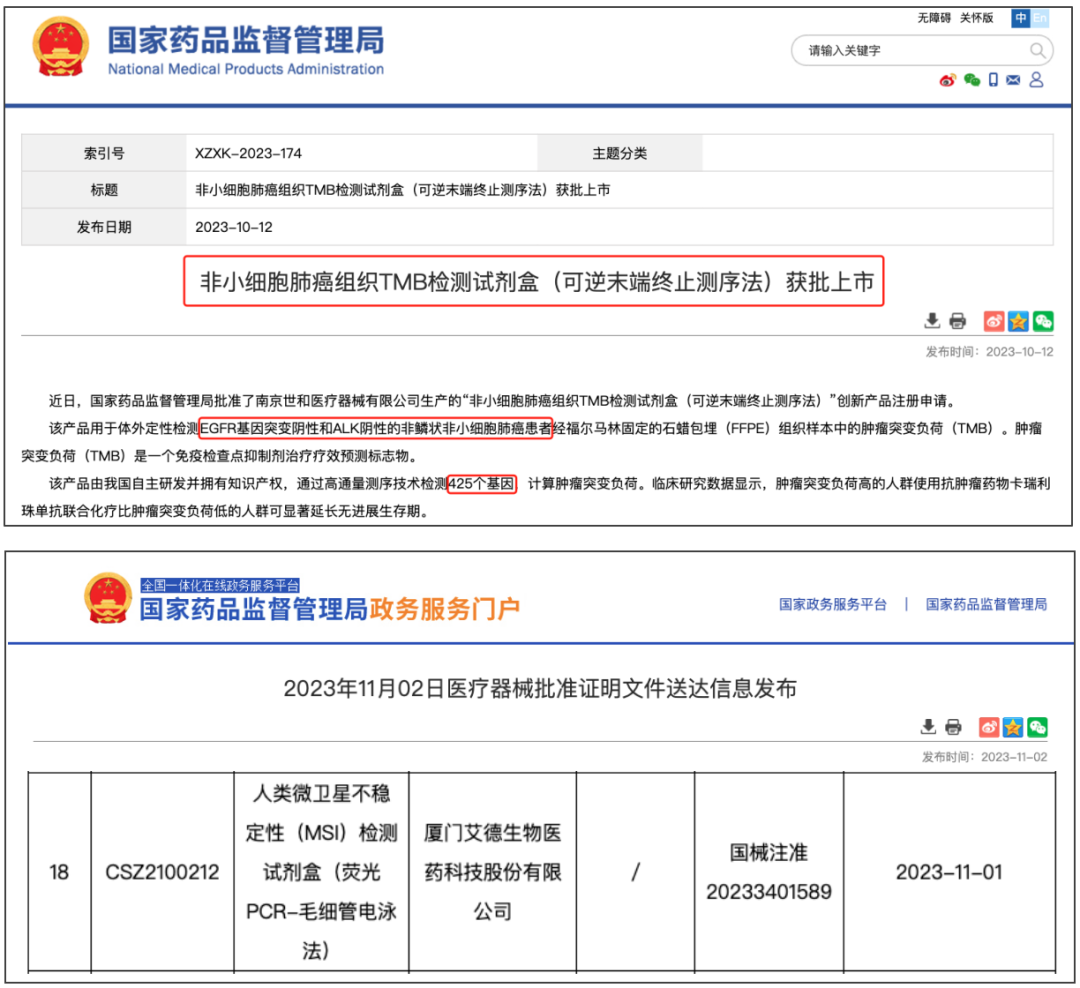
今年下半年,业内关于“规范基因检测外送”、“医疗反腐”的消息不绝于耳,整个基因检测行业式微的讨论甚嚣尘上。10月以来,世和TMB、艾德MSI获批的消息先后刷屏,给行业注入了两剂“强心针”,一扫眼下低迷之势。
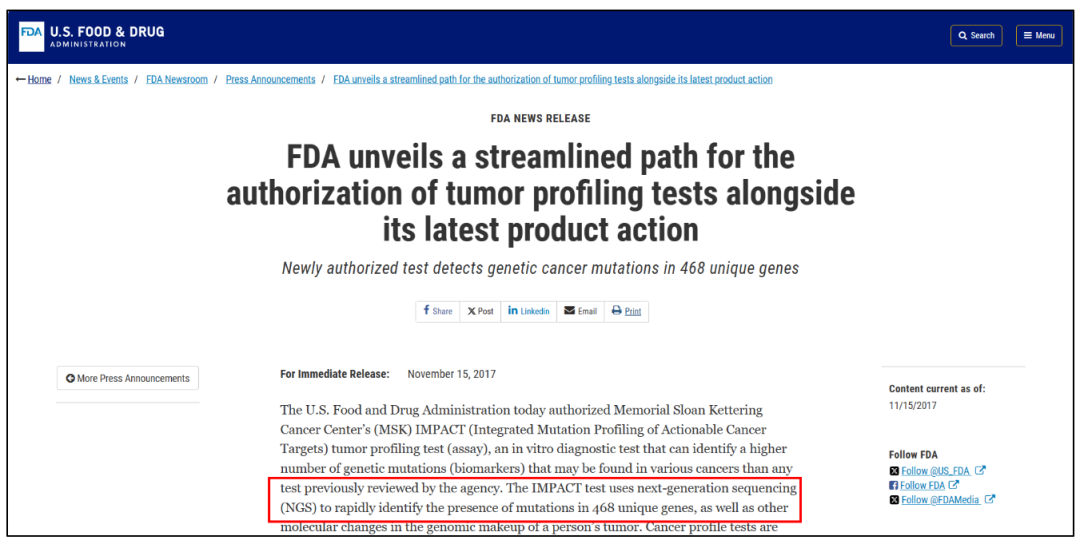
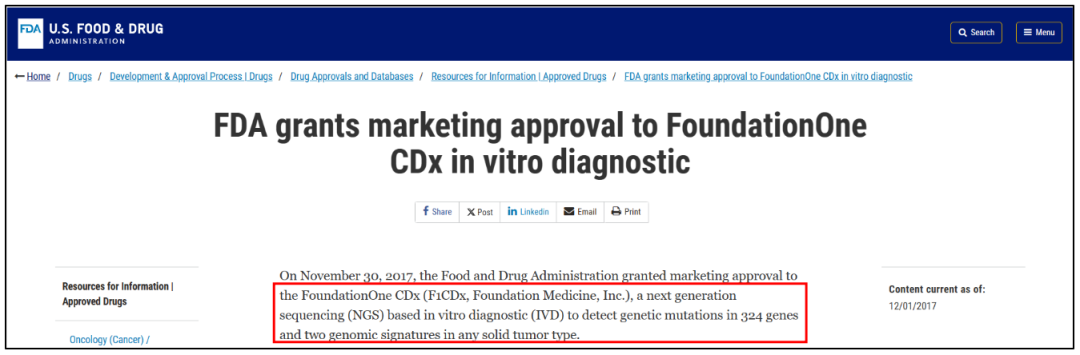
前者被称为NMPA批准的国内首个肿瘤NGS大panel产品,后者被誉为国内首个泛癌种免疫治疗伴随诊断试剂盒。免疫治疗发展史的2个重要的基因组标志物——TMB、MSI陆续以与“免疫治疗”关联的形式获批,值得业界鼓舞。 纵观国内医疗器械审批史,尤其是基因检测相关产品的审批历程,短短一个月内连续两款肿瘤免疫治疗相关试剂盒获批,且创造了多个“国内首个”,无疑称得上是行业里程碑事件。 这一幕似曾相识。 6年前,远在大洋彼岸的FDA,也曾在一个月内连续批准了两款重磅产品:
但两相比较之下,我们还是感受到了“中国首个”和“全球首个”之间的强烈落差。 落差产生的原因,不仅在产品本身,更在两地的审批逻辑。 “中国首个” VS “全球首个” 先粗略看一下NMPA和FDA分别批准的两款“首个”产品的关键指标:
单一指标、严格的适应征,也大大缩窄了产品的应用范围,这样“自砍几刀”,或许并非厂家申报之初的原意,而是在当前审批规则下尽快拿证的妥协之举。 反观FDA: MSK-IMPACT:检测与癌症相关的505个基因上的所有蛋白质编码突变、拷贝数变异、启动子突变以及结构重排的信息(获批时468个基因,后续更新到505个),多癌种,包括靶向疗法+免疫治疗疗效预测(含MSI)。 F1CDX:检测324个基因的 SNVs,Indels,CNVs和 Rearrangements 四种变异类型及TMB、MSI和HRD三种基因组特征,唯一一个拿下三个泛实体瘤药物治疗生物标志物(NTRK1/2/3,TMB,MSI-H)的伴随诊断产品。 一言以蔽之:不管是MSK-IMPACT,亦或是F1CDX,在6年前就同时实现了TMB、MSI和多个突变位点的“一揽子获批”。 所以,横向对比中美两个NGS产业发展最迅速的国家试剂盒审批史,我们在肯定并津津乐道于“首个”大panel在提振行业信心、叩响合规时代大门的同时,也应该思考一下,真正的NGS“大”panel,路在何方? 因为PCR1+PCR2+PCR3, 所以NGS1+NGS2+NGS3? 在反复研读上述两个国内“首个”产品的新闻时,一篇关于MSI获证的报道措辞颇耐人寻味。它是这样介绍该产品的临床价值和意义:
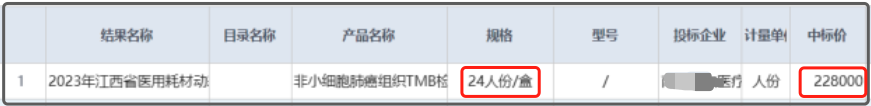
(来源:中国证券报) 该试剂盒的获批,“与......基因联检,实现了.......的合规检测,实现了......全病程管理”。简言之,这家幸运的企业,在拿到1个、2个、3个、4个证以后,终于才能实现对结直肠癌患者的全病程管理闭环。 以原有注册审批的通行做法,做一个基因位点验证一个基因位点,以连续性的位点攻略和覆盖,来实现某单癌种的全病程管理,这套逻辑毋庸置疑,在PCR领域很合理。原因也简单:PCR一次可以检测的基因位点有限。 正因如此,这种1+1+1最终达成效果3的产品申报和应用模式,就很PCR,而且这套规则在业内潜移默化了很多年,甚至变成了业内审评的一种惯性动作。 那么,在NGS领域呢?是不是也要沿用这套惯性动作,来一个,验证一个,批一个? 这很大程度上取决于NGS技术的特点。NGS之所以能占据一壁江山甚至后来居上,其最大的优势便是通量大,一次检测成千上百个基因位点不在话下,而且免疫标志物TMB、MSI等也尽收其中。可以说,一款NGS产品,足以满足临床检测对覆盖癌种、检测全面性的所有需求。 正因具备这样的特点,按理来说,对NGS产品的申报和应用模式,依其技术优势而言,在大方向上应该是“NGS=PCR1+PCR2+PCR3+……”这样的模式。 可惜的是,严格意义上讲,目前我们尚未看到一款这样“大而全”的产品。即便是让人喜大普奔的TMB试剂盒,按说明书严格执行,400多个基因仅能报告一个TMB指标,依然落入“NGS(TMB)+NGS1+NGS2+……”的窠臼。而且,仅TMB试剂盒挂网价格就高达9500元/人份,患者负担重,对医疗资源又何尝不是一种浪费?
对检测范围和应用范围的限制,也意味着需要更多的检测项目来满足某一患者的临床检测需求,即“联检”。看似单一项目收费降低,但“联检”的综合成本不容小觑。 与之对应的是,不同于PCR一个位点一个位点计费的传统模式,NGS一次检测几十个甚至数百个基因,得益于NGS全产业链上下游的快速发展,尤其是国产化测序平台的突破,NGS的检测成本已快速下探。 所以,如果我们沿用“PCR1+PCR2+PCR3”的组合申报模式,从而推演出“NGS1+NGS2+NGS3”的申报模式,对NGS技术而言,是扬短避长;对一款临检产品而言,是将“一个产品”可以解决的问题,人为切割为“多个产品”,这反而会带来社会医疗总成本的增加,受检者也无法从中获益。 NGS的发展应遵循自己的审批逻辑,才不会戴着镣铐跳舞。 三级位点,有望成为“跃龙门”的新路径吗? 如上所述,NGS的产品逻辑和申报现况的确还停留于“旧瓶装新酒”的阶段。 经过十多年发展、肿瘤NGS行业已经收获了近20个小panel试剂盒证,也算是小有所成。就在前不久的9月,中国器审发布了一篇《基于高通量测序技术(NGS)的肿瘤基因变异检测伴随诊断试剂的检测范围可以包括哪些基因及位点》一文,对一级、二级位点进行了明确说明,也借此解答了很多厂家提出的类似疑问。
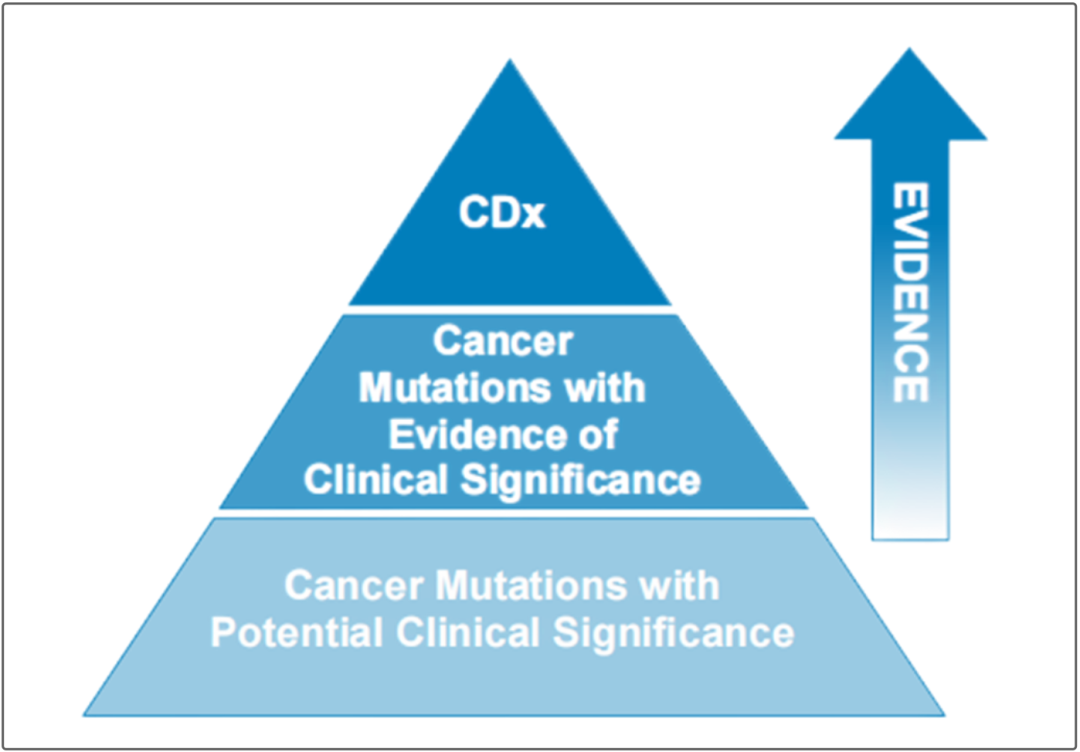
值得注意的是,除了严格定义“具有明确伴随诊断意义的基因和位点”为一级位点,文章将“尚无明确伴随诊断意义,但权威指南提出具有临床意义,且临床医生可以有依据的应用”的基因突变类型列为了二级位点。 但,这些对NGS大panel的价值而言,还远远不够。 很明显,如果检测范围只能是伴随诊断的一级和二级位点的数个基因,一方面与NGS高通量的产品特征不符;另一方面,这等于将未来升级适应症的路径也堵住了。 那么,升级的路径,还有更好的形式吗?或许,三级位点的设立是一种选择。 如果一个试剂盒里面,具有明确伴随诊断价值的一级和二级位点是名正言顺的“参赛选手”,那么,除了正式的参赛选手,是否可以考虑增加“三级位点”作为“候补选手”? 比如,按照FDA前几年发布的针对NGS检测中可报告生物标志物的三层金字塔式分类法,越往上,证据等级越高。
(https://www.fda.gov/media/109050/download) 我们不妨再来看一看国外同行的做法。 以FDA授权获批的OncoMine产品为例。(需要强调的是,OncoMine是一个可以售卖的kit,而不是single site assay。) 在2017年首次获批时,它的检测范围是非小细胞肺癌相关的23个基因,除具有伴随诊断价值的3个基因外,其他“AKT1、ALK、CDK4、DDR2……”20个基因都等同于“候补选手”。
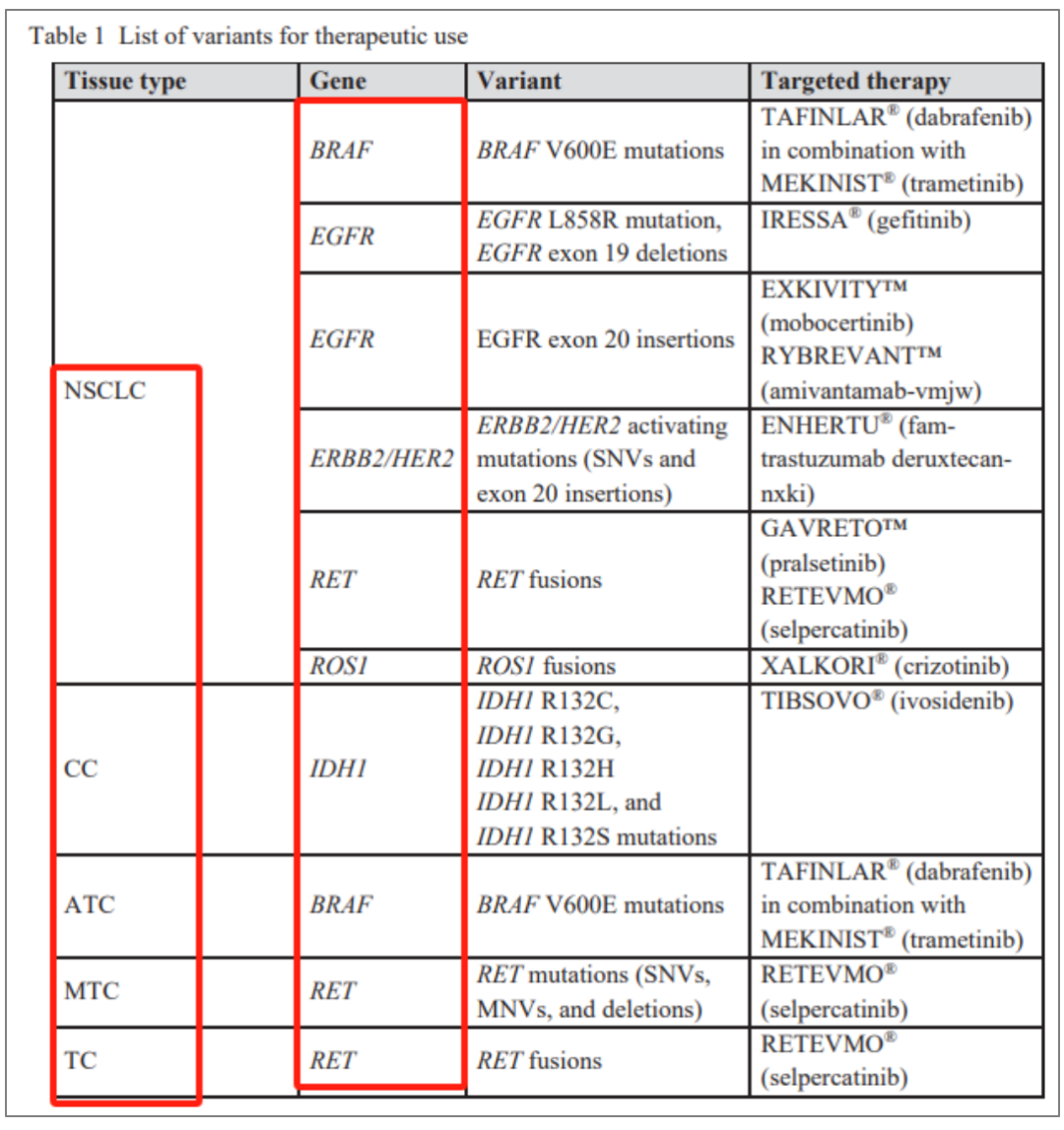
目前获批情况汇总:
到2023年11月,该产品检测范围已经扩展到了5个癌症类型的6个基因,而其实际检测的基因数远不止23个。 值得一提的是,在新增的胆管癌适应症扩展中(P160045/S028),产品本身存在的IDH1并没有出现在首次申报的23个基因行列,而是通过升级行为,IDH1从“未披露”一跃成为I级位点。
胆管癌IDH1获批情况 这样来看,OncoMine已经一步步展示了NGS试剂盒中“三级位点”存在的合理性和升级的可能性。结合这一操作实践和实际应用,我们姑且将三级位点的范围定义为: (1)具有潜在临床价值的基因位点; (2)符合NGS的技术特性,不会带来伴随诊断基因的性能受损和成本增加; (3)能与医药公司配合伴随诊断研究,节约开发成本并避免重复开发; 回过头来仔细想一想,一个体外诊断试剂盒从研发到注册,周期通常长达5-7年,而且整个过程所投入的研发和注册资金更是非常庞大。如果对一个NGS试剂盒检测范围的定型仅仅囿于当下的研究证据,而忽略了NGS技术本身的优势和成本特性,让最终获批的NGS体外诊断产品只能发挥出PCR体外诊断产品的效能,创新的意义何在? 因此,我们应当充分认识到NGS技术的潜力,以确保产品的获批不仅是时间和资金的投入,更是对患者医疗需求的全面回应。 写在最后 本篇从国内谈到国外,从PCR谈到NGS,无非是想业内对NGS大panel的定位拉齐到一个更符合其产品逻辑和应用场景的点,最朴素的理解,即“一次检测提供全面的、有价值的临床信息”。 我们也深知,在当今NGS成本已不再是其检测范围掣肘的时候,行业需要突破的是对超出规定范围外检测位点超预期用途的基因信息管理问题。 但不管怎么说,国内首个TMB注册证,至少在“可检测”、“可报告”、“可升级”的三个大panel合规化注册“难题”面前,已经勇敢地迈出了第一步。(详见:TMB第一证的“补充解读”:不去指鹿为马,才能预见未来)而对于真正基于伴随诊断用途的多基因位点或大panel,还需要继续突破这一限制。对此,企业、审批方、监管方、甚至患者的悲欢似乎并不相通。 如何破解这一问题? 引用之前业内资深人士的看法:“这件事,需要企业、上市审批和临床应用监管三方在互相信任的基础上一起来讨论、制订规则,去正面面对并找到降低‘超预期用途’的风险。” 经过这些年的发展,NGS技术在肿瘤基因检测上呈现出的临床价值已有目共睹,应用趋势也势不可挡。 相信在未来,科学监管与行业创新互为驱动,这些问题终将被解决! |


微信公众号

微博账号

商务合作